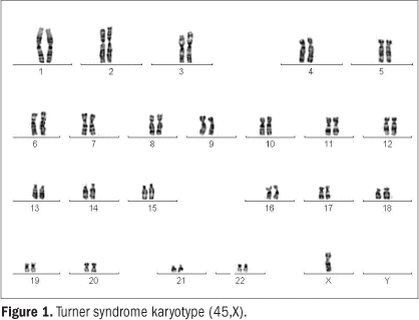
 A chromosome picture (karyotype) from a woman
with Turner syndrome. In this cell, the number of chromosomes is
45 with only one copy of the X chromosome (45,X) though other
cells in her body may have had the correct chromosome number
(46,XX)
A chromosome picture (karyotype) from a woman
with Turner syndrome. In this cell, the number of chromosomes is
45 with only one copy of the X chromosome (45,X) though other
cells in her body may have had the correct chromosome number
(46,XX)
Abnormalities of sex chromosomes can involve errors in the number of sex chromosomes, such as 45,X0 (Turner syndrome), 47,XXX, 47,XXY (Klinefelter syndrome), 47,XYY or mosaicism with at least one cell line having an aberrant number of sex chromosomes. Sex chromosome abnormalities also include aberrations of a single gene of the sex chromosome; resulting in errors in sex differentiation. This may result in 46,XX testicular disorder of sex development (DSD) 46,XY gonadal dysgenesis, 46,XX gonadal dysgenesis or ovatesticular DSD. (Claude J Migeon, 2007)
TURNER SYNDROME:
BACKGROUND
In 1938, Henry Turner described a group of female adolescents with primary amenorrhea, sexual infantilism and short stature. The patients also presented cubitus valgus, webbed neck, widely spaced nipples, low posterior hairline and lymphedema of hands and feet. Four years later, Varney and Albright et al.,independently from each other, studied patients with clinical signs resembling those described by Turner and demonstrated that, after puberty, they presented a high level of urinary gonadotropins, thus establishing that there was an abnormality of gonadal function rather than a hypothalamic or hypophyseal deficiency. In l944, Wilkins and Fleischmann performed histological analyses and observed that patients with the clinical signs described by Turner, Varney and Albright probably had streak gonads, and that all those authors were studying the same syndrome: Turner's syndrome. ( Rose Mary Rocco de Oliveira, Nov 2009)
INTRODUCTION
Turner's syndrome, a disorder in females characterized by the absence of all or part of a normal second sex chromosome, leads to a constellation of physical findings that often includes congenital lymphedema, short stature, and gonadal dysgenesis.Turner's syndrome occurs in 1 in 2500 to 1 in 3000 live-born girls. Approximately half have monosomy X (45,X), and 5 to 10 percent have a duplication (isochromosome) of the long arm of one X (46,X,i(Xq)). Most of the rest have mosaicism for 45,X, with one or more additional cell lineages. Some females with Turner syndrome have some cells with the usual 46,XX pattern and other cells will have 45,X. These individuals are said to be mosaic for Turner syndrome ( Virginia P. Sybert, 2004)
GENETIC CHARACTERISTICS
Cytogenetically, the Turner syndrome is characterized by sex chromosome monosomy (45,X) in phenotypically female individuals. This karyotype is found in 50-60% of the cases.The other cases are mosaics with a 45,X cell line accompanied by others with two or more X chromosomes or with structural anomalies. Such structural anomalies of the X chromosome (isochromosomes of the long arm, dicentric chromosomes, deletion of the short arm or ring chromosomes) are present in approximately 30% of the cases, in homogeneous karyotypes or in mosaics that include a 45,X cell line. Finally, around 5% are accounted for by patients with structural abnormalities of the Y chromosome (isochromosomes of the long arm and dicentric chromosomes) and mosaics that include a cell line accompanied by others with at least one Y chromosome, whether complete or not.
Data from the literature show that a second sex chromosome is necessary for the fetus to survive, and therefore virtually every liveborn 45,X individual should present more than one cell karyotype line, thus constituting a mosaic.This condition would be necessary for at least some organs, during a certain period of embryogenesis. This hypothesis is based on two main points: the frequency of sex chromosome mosaicism is much higher in liveborn infants with TS than in aborted fetuses; and an estimate that approximately 99% of the embryos with a pure 45,X karyotype die in utero.
Furthermore, comparison with other numerical chromosomal anomalies, such as Down syndrome or chromosome 18 trisomy, reveals that the incidence of TS does not increase with maternal age.These findings argue against meiotic nondisjunction as the main mechanism for the generation of a 45,X karyotype. Clinically, in contrast to other chromosomal syndromes, even patients with a pure 45,X karyotype present completely distinct phenotypes. Except for short stature, which seems to be a general characteristic, all other findings are inconsistent. One of the possible explanations for this fact might be undetected mosaicism, since the diagnosis is usually made by analyzing between 5 and 30 peripheral blood lymphocytes to determine the karyotype, and the second cell line is often present as a proportion of no more than 1 to 2% of the individual's cells.
In around 99% of human conceptuses with a 45,X karyotype, natural loss occurs during the first stage of embryonic development. Only 1% of these fertilizations are successful, and they generally display TS characteristics. Both embryonic mortality and the characteristic TS phenotype are considered to result from monosomy of genes that are common to the X and Y chromosomes (pseudoautosomal region). In women, it is clear that these genes are expressed both in the active and in the inactivated X chromosome, as a means of ensuring the proper amount of gene product. It is believed that one or more of these genes are responsible for TS.
DIAGNOSIS
When Turner's syndrome is diagnosed prenatally, the diagnosis is usually based on the finding of fetal edema on ultrasonography; abnormal levels of human chorionic gonadotropin, unconjugated estriol, and alpha-fetoprotein on screening of maternal serum (triple screening); or abnormal results of fetal karyotyping performed because of advanced maternal age. Affected fetuses often abort spontaneously. A 45,X fetus identified prenatally and surviving to birth has a prognosis similar to that of a child in whom Turner's syndrome is diagnosed postnatally. In contrast, approximately 90 percent of fetuses in whom 45,X/46,XX or 45,X/46,XY mosaicism is diagnosed incidentally during the course of screening for advanced maternal age or maternal triple screening will likely have a normal phenotype, female or male, respectively, at birth. The risk of eventual gonadal failure in these children with mosaicism is unknown.In contrast, a child in whom 45,X/46,XX or 45,X/46,XY mosaicism is diagnosed after birth is usually identified because of phenotypic features suggestive of Turner's syndrome; such children have a prognosis similar to that for 45,X children.
TURNER SYNDROME:
BACKGROUND
In 1938, Henry Turner described a group of female adolescents with primary amenorrhea, sexual infantilism and short stature. The patients also presented cubitus valgus, webbed neck, widely spaced nipples, low posterior hairline and lymphedema of hands and feet. Four years later, Varney and Albright et al.,independently from each other, studied patients with clinical signs resembling those described by Turner and demonstrated that, after puberty, they presented a high level of urinary gonadotropins, thus establishing that there was an abnormality of gonadal function rather than a hypothalamic or hypophyseal deficiency. In l944, Wilkins and Fleischmann performed histological analyses and observed that patients with the clinical signs described by Turner, Varney and Albright probably had streak gonads, and that all those authors were studying the same syndrome: Turner's syndrome. ( Rose Mary Rocco de Oliveira, Nov 2009)
INTRODUCTION
Turner's syndrome, a disorder in females characterized by the absence of all or part of a normal second sex chromosome, leads to a constellation of physical findings that often includes congenital lymphedema, short stature, and gonadal dysgenesis.Turner's syndrome occurs in 1 in 2500 to 1 in 3000 live-born girls. Approximately half have monosomy X (45,X), and 5 to 10 percent have a duplication (isochromosome) of the long arm of one X (46,X,i(Xq)). Most of the rest have mosaicism for 45,X, with one or more additional cell lineages. Some females with Turner syndrome have some cells with the usual 46,XX pattern and other cells will have 45,X. These individuals are said to be mosaic for Turner syndrome ( Virginia P. Sybert, 2004)
GENETIC CHARACTERISTICS
Cytogenetically, the Turner syndrome is characterized by sex chromosome monosomy (45,X) in phenotypically female individuals. This karyotype is found in 50-60% of the cases.The other cases are mosaics with a 45,X cell line accompanied by others with two or more X chromosomes or with structural anomalies. Such structural anomalies of the X chromosome (isochromosomes of the long arm, dicentric chromosomes, deletion of the short arm or ring chromosomes) are present in approximately 30% of the cases, in homogeneous karyotypes or in mosaics that include a 45,X cell line. Finally, around 5% are accounted for by patients with structural abnormalities of the Y chromosome (isochromosomes of the long arm and dicentric chromosomes) and mosaics that include a cell line accompanied by others with at least one Y chromosome, whether complete or not.
Data from the literature show that a second sex chromosome is necessary for the fetus to survive, and therefore virtually every liveborn 45,X individual should present more than one cell karyotype line, thus constituting a mosaic.This condition would be necessary for at least some organs, during a certain period of embryogenesis. This hypothesis is based on two main points: the frequency of sex chromosome mosaicism is much higher in liveborn infants with TS than in aborted fetuses; and an estimate that approximately 99% of the embryos with a pure 45,X karyotype die in utero.
Furthermore, comparison with other numerical chromosomal anomalies, such as Down syndrome or chromosome 18 trisomy, reveals that the incidence of TS does not increase with maternal age.These findings argue against meiotic nondisjunction as the main mechanism for the generation of a 45,X karyotype. Clinically, in contrast to other chromosomal syndromes, even patients with a pure 45,X karyotype present completely distinct phenotypes. Except for short stature, which seems to be a general characteristic, all other findings are inconsistent. One of the possible explanations for this fact might be undetected mosaicism, since the diagnosis is usually made by analyzing between 5 and 30 peripheral blood lymphocytes to determine the karyotype, and the second cell line is often present as a proportion of no more than 1 to 2% of the individual's cells.
In around 99% of human conceptuses with a 45,X karyotype, natural loss occurs during the first stage of embryonic development. Only 1% of these fertilizations are successful, and they generally display TS characteristics. Both embryonic mortality and the characteristic TS phenotype are considered to result from monosomy of genes that are common to the X and Y chromosomes (pseudoautosomal region). In women, it is clear that these genes are expressed both in the active and in the inactivated X chromosome, as a means of ensuring the proper amount of gene product. It is believed that one or more of these genes are responsible for TS.
DIAGNOSIS
When Turner's syndrome is diagnosed prenatally, the diagnosis is usually based on the finding of fetal edema on ultrasonography; abnormal levels of human chorionic gonadotropin, unconjugated estriol, and alpha-fetoprotein on screening of maternal serum (triple screening); or abnormal results of fetal karyotyping performed because of advanced maternal age. Affected fetuses often abort spontaneously. A 45,X fetus identified prenatally and surviving to birth has a prognosis similar to that of a child in whom Turner's syndrome is diagnosed postnatally. In contrast, approximately 90 percent of fetuses in whom 45,X/46,XX or 45,X/46,XY mosaicism is diagnosed incidentally during the course of screening for advanced maternal age or maternal triple screening will likely have a normal phenotype, female or male, respectively, at birth. The risk of eventual gonadal failure in these children with mosaicism is unknown.In contrast, a child in whom 45,X/46,XX or 45,X/46,XY mosaicism is diagnosed after birth is usually identified because of phenotypic features suggestive of Turner's syndrome; such children have a prognosis similar to that for 45,X children.
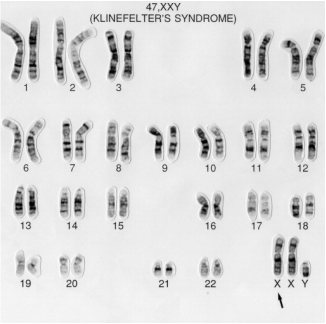
KLINEFELTER SYNDROME:
BACKGROUNG
Klinefelter syndrome 47,XXY was first described 70 years ago. With an incidence of 0.1% to 0.2% of male neonates (i.e. 1 to 2 per 1000), it is one of the commonest congenital chromosome disorders resulting in hypogonadism and genetically-determined infertility.(Eberhard Nieschlag, 2013)In 1942, Dr Harry Klinefelter published a report on nine men with a constellation of features: testicular dysgenesis, elevated urinary gonadotropins, microorchidism, eunuchoidism, azoospermia, and gynecomastia. It was believed to be an endocrine disorder of unknown etiology, until 1959, when Jacobs et al. recognized that Klinefelter syndrome was a chromosomal disorder in which there is an extra X chromosome resulting in the karyotype of 47,XXY. During the early 1970's, a number of centers began screening newborns for sex chromosomal abnormalities, because there was a need to obtain accurate information about childhood development in this condition . Previous studies of XXY individuals were extremely biased toward more severely affected individuals, since these patients were drawn largely from mental or penal settings where large numbers of men could be screened. These earlier studies implied a risk for mental deficiency and behavioral problems. As prospective, unbiased studied have reported their results in recent years, it has become clear that most XXY boys demonstrate reductions in speech and language abilities which are correlated with decreased reading and spelling achievement. Most, but not all XXY males, are infertile with small testicles, increased numbers of Leydig cells, tubular sclerosis, and interstitial fibrosis of varying degrees. Their ejaculate is usually azoospermic, and levels of testosterone are typically low to low-normal.
(Jeannie Visootsak and John M Graham, 2006)
INTRODUCTION:
The term Klinefelter syndrome describes a group of chromosomal disorder in which there is at least one extra X chromosome added to a normal male karyotype, 46,XY. The classic form is the most common chromosomal disorder, in which there is one extra X chromosome resulting in the karyotype of 47,XXY. As more individuals suspected of having Klinefelter syndrome had chromosome studies done, other karyotypes were sometimes observed, such as 48,XXYY; 48,XXXY and 49,XXXXY. (Jeannie Visootsak and John M Graham, 2006)
Klinefelter syndrome (KS) (47,XXY) occurs in ∼1 in 650 males, and is associated with a physical phenotype that can include tall stature, hypogonadism, and fertility problems. Although there have been many studies investigating the medical aspects of males with KS, it is also known that the presence of the extra X chromosome can lead to characteristic cognitive and language deficits of varying severity.
(Richard Boada, Jennifer Janusz, Christa Hutaff-Lee, and Nicole Tartaglia, 2009)
GENETIC CHARACTERISTICS
Klinefelter syndrome results from the presence of one extra copy of the X chromosome in each cell (47,XXY). Extra copies of genes on the X chromosome interfere with male sexual development, often preventing the testes from functioning normally and reducing the levels of testosterone. Most people with an extra X chromosome have the features described above, although some have few or no associated signs and symptoms.
Some people with features of Klinefelter syndrome have more than one extra sex chromosome in each cell (for example, 48,XXXY or 49,XXXXY). These conditions, which are often called variants of Klinefelter syndrome, tend to cause more severe signs and symptoms than classic Klinefelter syndrome. In addition to affecting male sexual development, variants of Klinefelter syndrome are associated with intellectual disability, distinctive facial features, skeletal abnormalities, poor coordination, and severe problems with speech. As the number of extra sex chromosomes increases, so does the risk of these health problems. (Frühmesser A, Kotzot D, 2011)
Some people with features of Klinefelter syndrome have the extra X chromosome in only some of their cells; in these individuals, the condition is described as mosaic Klinefelter syndrome (46,XY/47,XXY). Individuals with mosaic Klinefelter syndrome may have milder signs and symptoms, depending on how many cells have an additional X chromosome. Mosaic individuals have an even wider diversity of findings, especially in the histologic characteristics of testicular biopsies.Samples from male subjects with XY/XXY genotype reveal that 14% to 61% of their seminiferous tubules contain maturing spermatids, which explains the rare cases of fertility in these patients. (Cynthia M. Smyth, MD; William J. Bremner;June 22, 1998)
DIAGNOSIS
The 'prototypic' man with Klinefelter syndrome has traditionally been described as tall, with narrow shoulders, broad hips, sparse body hair, gynecomastia, small testicles, androgen deficiency, azoospermia and decreased verbal intelligence. A less distinct phenotype has, however, been described. Klinefelter syndrome is an underdiagnosed condition; only 25% of the expected number of patients are diagnosed, and of these only a minority are diagnosed before puberty. ( Bojesen A, Gravholt CH, 2007)
BACKGROUNG
Klinefelter syndrome 47,XXY was first described 70 years ago. With an incidence of 0.1% to 0.2% of male neonates (i.e. 1 to 2 per 1000), it is one of the commonest congenital chromosome disorders resulting in hypogonadism and genetically-determined infertility.(Eberhard Nieschlag, 2013)In 1942, Dr Harry Klinefelter published a report on nine men with a constellation of features: testicular dysgenesis, elevated urinary gonadotropins, microorchidism, eunuchoidism, azoospermia, and gynecomastia. It was believed to be an endocrine disorder of unknown etiology, until 1959, when Jacobs et al. recognized that Klinefelter syndrome was a chromosomal disorder in which there is an extra X chromosome resulting in the karyotype of 47,XXY. During the early 1970's, a number of centers began screening newborns for sex chromosomal abnormalities, because there was a need to obtain accurate information about childhood development in this condition . Previous studies of XXY individuals were extremely biased toward more severely affected individuals, since these patients were drawn largely from mental or penal settings where large numbers of men could be screened. These earlier studies implied a risk for mental deficiency and behavioral problems. As prospective, unbiased studied have reported their results in recent years, it has become clear that most XXY boys demonstrate reductions in speech and language abilities which are correlated with decreased reading and spelling achievement. Most, but not all XXY males, are infertile with small testicles, increased numbers of Leydig cells, tubular sclerosis, and interstitial fibrosis of varying degrees. Their ejaculate is usually azoospermic, and levels of testosterone are typically low to low-normal.
(Jeannie Visootsak and John M Graham, 2006)
INTRODUCTION:
The term Klinefelter syndrome describes a group of chromosomal disorder in which there is at least one extra X chromosome added to a normal male karyotype, 46,XY. The classic form is the most common chromosomal disorder, in which there is one extra X chromosome resulting in the karyotype of 47,XXY. As more individuals suspected of having Klinefelter syndrome had chromosome studies done, other karyotypes were sometimes observed, such as 48,XXYY; 48,XXXY and 49,XXXXY. (Jeannie Visootsak and John M Graham, 2006)
Klinefelter syndrome (KS) (47,XXY) occurs in ∼1 in 650 males, and is associated with a physical phenotype that can include tall stature, hypogonadism, and fertility problems. Although there have been many studies investigating the medical aspects of males with KS, it is also known that the presence of the extra X chromosome can lead to characteristic cognitive and language deficits of varying severity.
(Richard Boada, Jennifer Janusz, Christa Hutaff-Lee, and Nicole Tartaglia, 2009)
GENETIC CHARACTERISTICS
Klinefelter syndrome results from the presence of one extra copy of the X chromosome in each cell (47,XXY). Extra copies of genes on the X chromosome interfere with male sexual development, often preventing the testes from functioning normally and reducing the levels of testosterone. Most people with an extra X chromosome have the features described above, although some have few or no associated signs and symptoms.
Some people with features of Klinefelter syndrome have more than one extra sex chromosome in each cell (for example, 48,XXXY or 49,XXXXY). These conditions, which are often called variants of Klinefelter syndrome, tend to cause more severe signs and symptoms than classic Klinefelter syndrome. In addition to affecting male sexual development, variants of Klinefelter syndrome are associated with intellectual disability, distinctive facial features, skeletal abnormalities, poor coordination, and severe problems with speech. As the number of extra sex chromosomes increases, so does the risk of these health problems. (Frühmesser A, Kotzot D, 2011)
Some people with features of Klinefelter syndrome have the extra X chromosome in only some of their cells; in these individuals, the condition is described as mosaic Klinefelter syndrome (46,XY/47,XXY). Individuals with mosaic Klinefelter syndrome may have milder signs and symptoms, depending on how many cells have an additional X chromosome. Mosaic individuals have an even wider diversity of findings, especially in the histologic characteristics of testicular biopsies.Samples from male subjects with XY/XXY genotype reveal that 14% to 61% of their seminiferous tubules contain maturing spermatids, which explains the rare cases of fertility in these patients. (Cynthia M. Smyth, MD; William J. Bremner;June 22, 1998)
DIAGNOSIS
The 'prototypic' man with Klinefelter syndrome has traditionally been described as tall, with narrow shoulders, broad hips, sparse body hair, gynecomastia, small testicles, androgen deficiency, azoospermia and decreased verbal intelligence. A less distinct phenotype has, however, been described. Klinefelter syndrome is an underdiagnosed condition; only 25% of the expected number of patients are diagnosed, and of these only a minority are diagnosed before puberty. ( Bojesen A, Gravholt CH, 2007)
This website is designed for educational purpose only.
